Methodology
This page contains details on the methodologies used to generate the data on this website.
IMPORTANT
If you are interested in using data from these experiments, we recommend that you read the entirety of this page. We also suggest that you pay special attention to the Data format and commonly used filters section as a guide to understanding the raw data.
Overview
Deep mutational scanning (DMS) refers to the high-throughput measurement of all or nearly all mutations in a protein of interest.
Typically, DMS involves:
- Generating a pool of genotype-phenotype linked protein mutants.
- Imposing a selection on the pool of mutants based on your phenotype of interest.
- Measuring the change of frequency of each mutant protein with deep sequencing.
DMS on viral entry proteins (VEPs) is performed by producing a pool of viruses that contains nearly all possible VEP mutations or combinations of mutations. A selection is applied to this pool of viruses by attempting to infect susceptible cells. Viruses with mutant VEPs capable of accomplishing cell entry are enriched. Additionally, the pool of mutant viruses can be incubated with antibodies, serum, or other interacting proteins before infecting cells. In this case, viruses with mutant VEPs that abrogate binding by antibodies or other interacting proteins are enriched.
In the past, a subamplicon sequencing strategy was used to measure the change in frequencies of mutations across selections in VEP DMS experiments. However, experimental challenges, biosafety concerns, and the inability to simultaneously measure the effects of combinations of mutations led the Bloom lab to develop a lentiviral pseudotyping approach to VEP DMS.
A lentivirus-based DMS platform
In the Bloom lab, we've developed a lentivirus-based approach for performing DMS on any VEP that can be pseudotyped with a lentiviral vector. Our approach uses controlled lentivirus replication cycles and a barcode sequencing strategy to enable the measurement of individual or combinations of mutations in viral mutants within the pool.

The key features in the lentiviral vector that enable deep mutational scanning are the random nucleotide barcode following the VEP (in this example, HIV Envelope) and the repaired 3' LTR. The repair of the 3' LTR enables the lentivirus to undergo multiple controlled cycles of replication rather than only a single cycle. The barcode following the VEP enables the use of PacBio long-read sequencing to link mutants with their unique barcode so that only Illumina short-read sequencing of the barcodes is needed in downstream experiments to measure mutant frequencies. The VEP is also under an inducible promoter, so the mutant VEP library is not expressed before it is induced with doxycycline during genotype-phenotype linked virus production.
Making a genotype-phenotype linked library is a two-step process:
- The VSV-G viral entry protein is used to integrate the lentivirus genomes containing the VEP mutants and barcodes into cells at just one copy per cell.
- The genotype-phenotype linked viruses are produced from singly integrated cells.
Viruses can also be produced from the singly integrated cells separately with VSV-G added. These VSV-G pseudotyped viruses can be used to infect cells regardless of mutant VEP function to determine the library's composition of genotype-phenotype linked mutants by long-read sequencing. Finally, this library can be used for a variety of downstream experiments involving different selections.
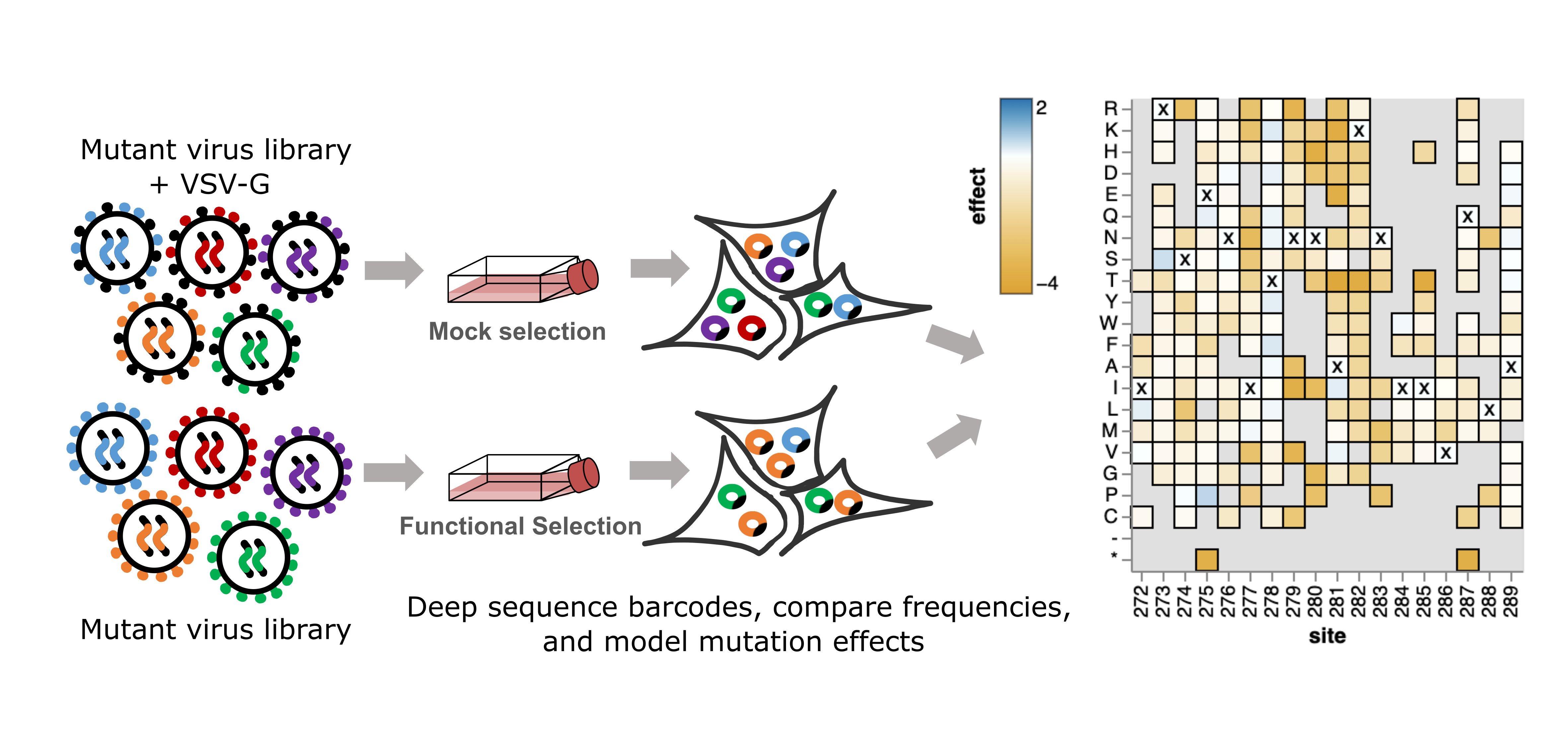
Measuring the effects of mutations on cell entry
Once the linkage between viral mutant and barcode has been determined by long-read sequencing, the experiments to determine the effects of mutations on cell entry are straightforward. The mutant library is produced from the singly integrated cells with and without the addition of VSV-G. Both VSV-G + and - pools are used to infect susceptible cells separately. The change in frequency of the mutants that successfully infect cells between conditions is then used to score each mutation based on its effect on cell entry. A statistical model (detailed here) is used to deconvolute the effects of individual mutants.
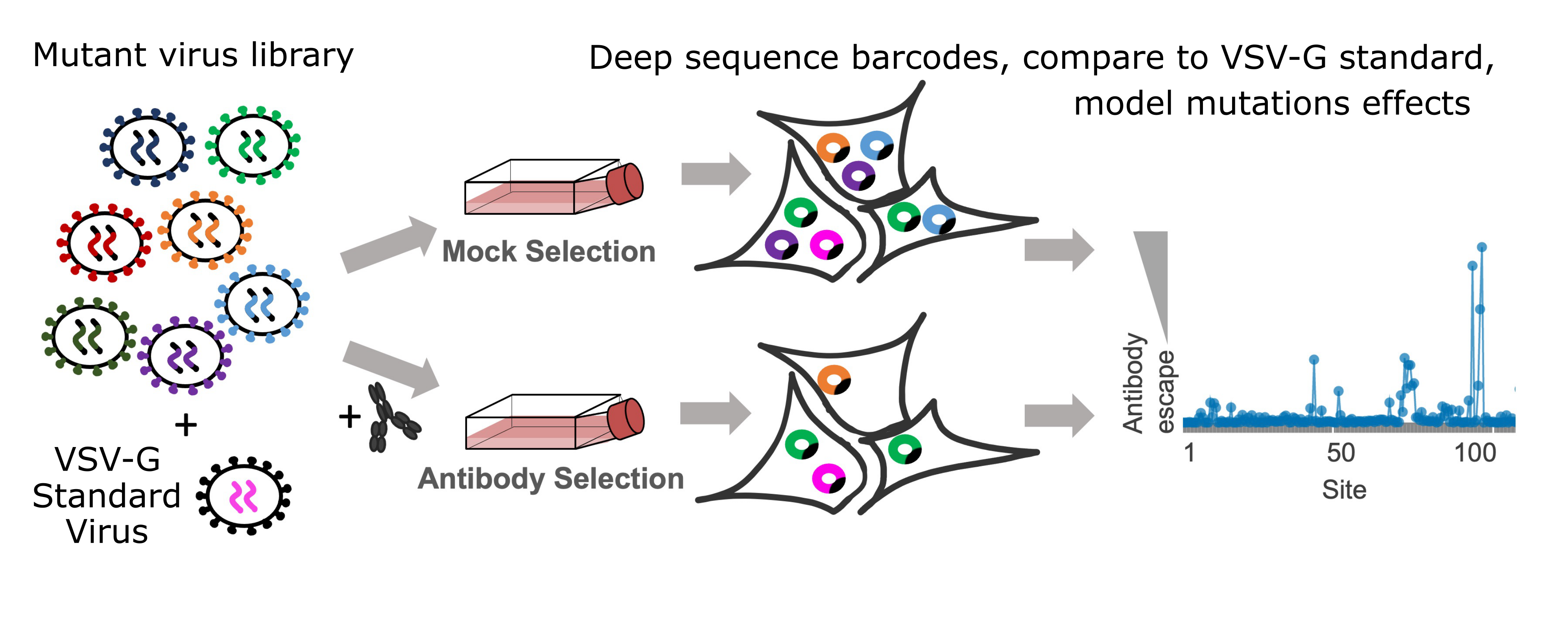
Measuring the effects of mutations on escape from neutralization
To determine the effect of mutations on antibody or serum escape, an additional step is involved. A small amount of virus pseudotyped with only VSV-G and a set of known barcodes is spiked into the library. The mutant virus library containing the VSV-G standard spike-in is split into several conditions including a mock condition without antibodies or serum and several conditions with varied concentrations of antibodies or sera. Since the VSV-G standards are not neutralized by the antibodies or sera, the extent to which each viral mutant is neutralized is determined by comparing its frequency in the VSV-G standard with its frequency in the antibody or serum conditions. These escape scores are then used to fit a biophysical model where each mutation affects neutralization at one or more epitopes for each antibody or serum. See the Modeling the effects of mutations on escape from antibodies and sera section in the computational methods section below for details on this modeling.
Measuring the effects of mutations on receptor binding
To determine the effects of mutations on receptor binding, we use a similar approach to experiments that measure the effect of mutation on neutralization escape. Instead of incubating the viral library with antibodies or serum, the library is incubated with soluble receptor protein. The VEPs bound by soluble receptors will be blocked from binding cells. By incubating the mutant virus library with a wide range of soluble receptor concentrations, we can measure the effects of mutations that increase or decrease soluble receptor binding.
Computational methods
We rely on modeling to calculate scores for each mutant (or their combination) that reflect their impact on cell entry, antibody escape, or receptor binding. The sections below provide a high-level overview of the computational modeling used to calculate these scores.
Modeling the effects of mutations on cell entry
To deconvolve the effects of individual VEP mutations on cell entry, we use global epistasis models. Global epistasis models work by defining a latent, unmeasured scale of mutation effects where mutations have an additive effect on function. This latent scale can be transformed into the observed, measured scale of the effects of mutations using a non-linear function, typically similar to a sigmoid. Through these models, a significant portion of the epistasis, or non-additive effects of combinations of mutations, can be modeled as the non-linearity between the latent and observed scales. To fit this non-linearity in global epistasis models, there must be measurements of mutants with combinations of mutations, which is made possible with the lentiviral vector DMS platform.
The functional effects of mutations reported in publications and figures are typically the predicted observed effect of each mutation when present by itself unless it is labeled as the latent effect. When the latent effect is reported, it is the predicted effect of that mutation on the additive, latent scale.
Modeling the effects of mutations on escape from antibodies and sera
To deconvolve the effects of individual mutations on VEP escape from neutralization by antibodies or sera, we fit a biophysical model where the level of neutralization at a particular concentration of antibodies or serum is determined by neutralizing activity at one or more epitopes, along with the concentration of antibodies or serum. Under this model, individual mutations have additive effects on the free energy of binding at each epitope, and the summed effects of mutations can be transformed into the fraction of virus bound at each epitope using a non-linear function, similar to global epistasis models. The product of the fraction unbound at each epitope then determines the total fraction of unbound virus.
The escape scores of the effects of mutations at each epitope reported in publications and figures are typically the additive effect of each mutation on the modeled free energy of binding at that epitope. These effects can be used to predict the concentrations of antibody or serum necessary to achieve an arbitrary level of neutralization for a VEP mutant with one or more mutations (for example, an IC50 or IC80) using the fit biophysical model. These predictions are also often reported in publications to compare results to traditional neutralization assays.
Modeling the shifts in effects of mutations between strains
We also use models to compare the shifts in effects of mutations between stains of the same VEP. These models are essentially extensions of global epistasis models, where the latent effects of mutations between strains are compared. Since it is expected that most mutations will not have shifted effects between strains that aren't highly divergent, this approach includes regularization to force shifts toward zero unless there is high confidence in a shifted effect.
Data format and commonly used filters
This section describes common data formats and filters that will are important for exploring the raw DMS data. The following examples are taken from Carr, Crawford et at., (2024).
Data formats
Each study is accompanied by a GitHub repository and pages containing the data, analysis, and results for that study. Here is an example of a GitHub repository and its corresponding website hosted on GitHub pages. The GitHub pages site provides a more intuitively organized way to access the information on GitHub.
The least processed form of the data is the sequencing files deposited on the SRA and their corresponding configuration files in the GitHub repository. The repository can be cloned and run to perform the entire analysis. However, re-running the analysis isn't necessary for most applications.
The scores reflecting the raw effect of each mutation on cell entry (function scores) or antibody selections are stored as .csv files in the GitHub repository and are often also linked to in the GitHub pages site. Here is an example of a file containing functional effects scores for mutation in the Lassa glycoprotein. In these files, each row contains information about an individual mutant, such as its barcode, amino acid mutations, and score. You can use these files to do things like fit epistasis models, antibody escape models, or other models that require measurements of the effects of combinations of mutations.
The scores of mutants in individual selections are used by our analysis pipeline to fit epistasis and escape models (described above). We use dms-variants to fit global epistasis models and polyclonal to fit models of immune escape. The fit models themselves are large and typically not stored on GitHub. To fit your own versions of these models, you can use the scores for variants from individual selections and follow the examples in the dms-variants and polyclonal documentations.
After fitting these models, the predictions of the effects of mutations are averaged across selections, and then reported in .csv files along with some additional information. Here is an example of a file with average effects of mutations on function across models. Here is an example of a file with the average effects of mutations on antibody escape. For each mutation, there is information about the average score, the standard deviation of the score, and the times_seen for that mutation, which will be explained in the next section. We typically report these average or median scores in publications and figures.
Common filters across experiment types
There are several filters to understand if you want to use the raw DMS data.
One of the most important filters is the times_seen metric. 'Times seen' refers to the number of viral mutants in a selection that contain a particular mutation. If a mutation was observed in very few mutant contexts, we have less confidence in the estimated effect of that mutation. Therefore, we usually filter out mutations observed in fewer than three mutant contexts (times_seen < 3). As this filter is increased, the confidence in the effects of mutations increases, and correlations between replicates and independent mutant libraries will increase. However, the total number of mutations in the filtered dataset will decrease.
The standard deviation of scores is another important metric to consider for filtering data. When scores for a mutation differs greatly between selections or independent mutant libraries, it's a sign that the measurements for that mutation may be noisy or unreliable. This is not always true, since sometimes antibody escape mutations may have very high scores in one library but not as high but still positive scores in another library. Interactive plots made in the analysis pipeline will often display standard deviations and the scores for mutations for each independent mutant library or independent selections when moused over.
Experiment-specific filters
For antibody and serum escape data, it can also be useful to apply filters on mutations based on their effect on function of entry into cells. The reason for this is because poorly functioning mutants are often a very low frequency in the mock selection with no antibody, but can become a relatively high fraction of the counts just by chance in the antibody selection as most of the wildtype and mutant viruses are neutralized. By randomly going from a very low count to a relatively high fraction of the counts in some selections, these poorly functioning variants often have very noisy effects on escape from antibodies or sera. We often filter these mutations out of plots and figures by filtering out any mutations with a worse functional effect than some quantile (~.75) of stop codon effect.